Introducción
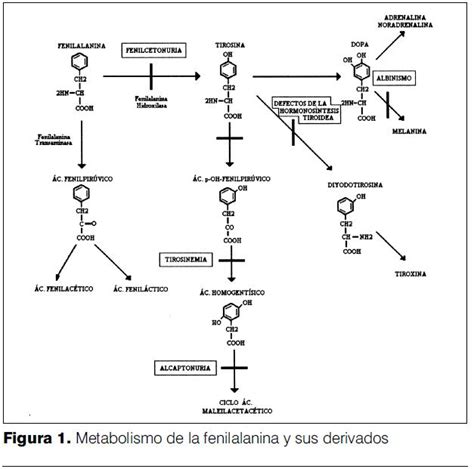
Las hiperfenilalaninemias (HFA) son un grupo de trastornos metabólicos que se originan por una deficiencia o ausencia de la enzima fenilalanina hidroxilasa (FAH). Esta enzima es crucial para el metabolismo de la fenilalanina (FA), convirtiéndola en tirosina (TIR). Cuando la FAH no funciona correctamente, la fenilalanina se acumula en la sangre y el cerebro, lo que puede causar un daño progresivo en el sistema nervioso central. Si la enfermedad no se diagnostica y trata a tiempo, puede derivar en un retardo mental profundo.
Un diagnóstico tardío de la fenilcetonuria (PKU) puede manifestarse a través de diversas alteraciones, incluyendo problemas conductuales, hipopigmentación de la piel y el cabello, convulsiones y un característico olor a humedad. Por otro lado, el diagnóstico neonatal y el inicio temprano del tratamiento nutricional han demostrado ser altamente efectivos en la prevención de secuelas neurológicas.
La herencia de la fenilcetonuria es autosómica recesiva, lo que implica que existe un riesgo del 25% de recurrencia en cada embarazo si ambos padres son portadores del gen defectuoso. En Chile, se ha identificado una frecuencia de la mutación IVS10 del 21.1%, similar a la encontrada en España.
Clasificación de las Hiperfenilalaninemias
Las HFA se clasifican según varios criterios, incluyendo el nivel de fenilalanina plasmática, la tolerancia a la ingesta de este aminoácido y la actividad enzimática residual:
- Fenilquetonuria (PKU) clásica: Los niveles de FA en plasma superan los 20 mg/dL (normalmente, son inferiores a 2 mg/dL), con niveles de TIR por debajo de 0.8 mg/dL (normalmente entre 1 y 1.5 mg/dL). Se observa la presencia de fenilcetonas en la orina y la actividad de la FAH es inferior al 1%. Estos pacientes tienen una tolerancia a la FA dietaria menor de 20 mg/kg/día.
- PKU moderada: Los niveles de FA en plasma oscilan entre 4 y 19 mg/dL, con niveles de TIR dentro del rango normal. La actividad de la FAH se sitúa entre el 3% y el 50%, y la tolerancia a la FA dietaria es de 20 a 25 mg de FA/kg/día.
- PKU leve: Presentan niveles séricos de FA de 4 a 10 mg/dL y toleran ingestas de FA superiores a 50 mg/kg/día.
- HFA leve: Los niveles de FA plasmática se encuentran entre 4 y 10 mg/dL, con niveles de TIR normales. La actividad de la FAH es mayor del 50% y tiende a normalizarse después de los 6 meses de edad. Estos pacientes no requieren de una dieta restrictiva.
Clínica y Diagnóstico
La sintomatología de la fenilcetonuria puede manifestarse desde los primeros meses de vida. Entre los síntomas iniciales se incluyen la falta de interés por el entorno, convulsiones, espasmos masivos con hipsarritmia en el electroencefalograma, eczema resistente al tratamiento y una coloración más clara del cabello, ojos y piel en comparación con los progenitores. Un signo distintivo es el olor a humedad, producido por la excreción de ácido fenilacético.
Alrededor de los 6 meses de edad, se hace evidente el retraso en el desarrollo psicomotor. En niños mayores, pueden aparecer graves trastornos de conducta como agresividad, hiperactividad, rabietas, conductas autodestructivas y actitudes autistas. En la edad adulta, los individuos con PKU pueden experimentar regresión intelectual y deterioro neurológico asociado a la desmielinización.
El aumento de la FA en la sangre interfiere con el transporte de otros aminoácidos aromáticos, dibásicos y neutros a través de las membranas celulares, incluyendo la barrera hematoencefálica. Esta alteración en el cerebro reduce las concentraciones de aminoácidos intraneuronales y afecta la síntesis proteica, la proliferación dendrítica temprana y la mielinización. Consecuentemente, se incrementa el reciclaje de mielina y se inhibe la síntesis de neurotransmisores como la serotonina, dopamina y norepinefrina.
Se ha demostrado de manera concluyente que el diagnóstico antes del primer mes de vida previene el daño neurológico. Los programas de detección neonatal para PKU, iniciados en los países desarrollados en 1963 mediante la prueba de Guthrie, han logrado erradicar esta patología como causa de retardo mental. La incidencia global estimada de PKU es de 1:10.000 recién nacidos vivos (RNV), con variaciones significativas entre grupos étnicos (por ejemplo, 1:110.000 RNV en Japón y 1:6.000 RNV en Turquía, debido a la alta consanguinidad). En Chile, la incidencia de PKU clásica se ha estimado en 1:21.509 RNV, y la de HFA en 1:14.416 RNV.
Se han identificado más de 500 mutaciones asociadas a la fenilcetonuria, y en el 79% de los casos existe una correlación entre el genotipo y el fenotipo. Estudios prospectivos en niños con PKU detectados precozmente que suspendieron la dieta entre los 6 y 12 años de edad, evidenciaron una disminución del coeficiente intelectual, cambios conductuales y déficit atencional, así como alteraciones en la mielinización detectadas por resonancia magnética cerebral. Sin embargo, estos síntomas revierten al reintroducir la dieta restrictiva.
Considerando estos hallazgos, se recomienda mantener la dieta durante toda la vida, especialmente en mujeres con PKU, debido al efecto teratogénico de la HFA sobre el feto. Niveles de FA superiores a 6 mg/dL durante el embarazo pueden provocar microcefalia, bajo peso al nacer (<2.500 g), cardiopatías congénitas y retardo mental en el feto. La instauración de un tratamiento dietético estricto antes de la concepción previene estas secuelas.
Tratamiento
El tratamiento de la fenilcetonuria se basa en una dieta restringida en fenilalanina (FA), que, al ser iniciada desde el período neonatal, previene el retardo mental. La dietoterapia consiste en limitar la ingesta de FA a 250-500 mg/día (la dieta normal aporta entre 3.000 y 5.000 mg/día). El objetivo principal es mantener los niveles de FA en sangre entre 2 y 10 mg/dL (120 - 600 uM/L), niveles que permiten un crecimiento y desarrollo normal en los niños con PKU.
El Consejo de Investigaciones Médicas (Medical Research Council) ha establecido que todo niño con PKU que presente niveles de FA superiores a 10 mg/dL con una dieta normal debe iniciar una dieta restringida en FA. Esta dieta excluye alimentos de origen animal, que son ricos en FA, y restringe el consumo de cereales, frutas y verduras.
Un sustituto lácteo especial sin FA es fundamental, ya que constituye la única fuente de proteínas de alto valor biológico para estos pacientes. Se han desarrollado diversos alimentos con bajo contenido proteico (fideos, galletas, pan, chocolates) que proporcionan calorías adicionales, promueven la saciedad y ayudan a prevenir transgresiones dietéticas en niños mayores.
Manejo Nutricional Inicial
En la primera semana de tratamiento, se suspende la lactancia materna y se administra el 100% del líquido requerido como fórmula especial sin FA (150 ml/kg/día). Las calorías se complementan con maltodextrina y aceite de soya, ajustadas según sexo, edad y peso real del niño.
Se monitorean semanalmente los niveles de FA en sangre mediante el método de inhibición bacteriana de Guthrie, y se ajusta la ingesta de FA en consecuencia. La prescripción inicial incluye la fórmula especial, seguida de la lactancia materna cada 3 horas.
Protocolo de Evaluación del Nivel de FA en Sangre con Lactancia Materna:
- Si el nivel de FA se encuentra entre 2 - 6 mg/dL: Se mantiene la prescripción.
- Si está bajo 2 mg/dL: Se reduce la fórmula especial en un 25%, aumentando indirectamente el volumen de leche materna y la cantidad de FA dietaria.
- Si el nivel de FA en sangre está entre 6 y 10 mg/dL: Se incrementa el volumen de la fórmula especial sin FA en un 25%, disminuyendo indirectamente la ingesta de leche materna.
- Si el nivel de FA se encuentra sobre 10 mg/dL: Se aumenta la fórmula en un 50% del total de líquidos prescritos, según el peso real del niño.
El Instituto de Nutrición y Tecnología de los Alimentos (INTA) de la Universidad de Chile ha incorporado la lactancia materna en el tratamiento de la PKU, observando que hasta el cuarto mes de edad, la leche materna aporta el 50% de las proteínas, calorías y FA, manteniendo los niveles de FA en sangre por debajo de 6.0 mg/dL. Al sexto mes, el 74% de los niños con PKU continúan recibiendo lactancia materna y fórmula especial.

Suplementación Nutricional y Otros Nutrientes
La dieta restrictiva por sí sola no asegura un aporte adecuado de nutrientes esenciales como zinc, selenio, hierro, cobre, cromo, vitamina B12 y calcio, por lo que es necesario suplementarlos farmacológicamente o a través de la fórmula especial sin FA.
Otros nutrientes que pueden estar en déficit son los ácidos grasos esenciales: araquidónico (AA) y docosahexaenoico (DHA). Dada su importancia en las estructuras de la retina y el cerebro, se recomienda mantener una relación entre linoleico y alfa linolénico de 1:15 para favorecer la síntesis de AA y DHA a partir de sus precursores.
Se ha reportado que niños con PKU en dieta estricta pueden presentar desmineralización ósea, aumentando el riesgo de fracturas. La deficiencia de tirosina (TIR) puede reducir la síntesis de dopamina y noradrenalina, ya que el transportador de aminoácidos neutros hacia la barrera hematoencefálica tiene mayor afinidad por la FA, limitando el ingreso de triptófano y tirosina. Por ello, se sugiere suplementar con TIR (100-300 mg/kg/día) para mantener sus niveles en sangre por encima de 0.8 mg/dL (50-100 uM/L).
Manejo en Situaciones Especiales
El aumento de la FA plasmática durante el seguimiento puede deberse a infecciones (catabolismo), ayuno prolongado, cirugías o transgresiones alimentarias. En estas situaciones, el tratamiento dietético consiste en disminuir la ingesta de FA en un 25%, aumentar el volumen de la fórmula sin FA para promover la síntesis proteica y incrementar la ingesta de energía, hasta lograr el balance metabólico deseado.
En la PKU materna, la dieta debe ser tan estricta como en los lactantes, ya que la FA atraviesa la placenta con una gradiente positiva, pudiendo alcanzar niveles fetales de 1 a 2.5 veces superiores a los maternos. Para prevenir el efecto teratógeno, se recomienda iniciar la dieta restringida en FA dos meses antes de la concepción y mantenerla durante todo el embarazo. La dieta debe aportar entre 6 y 10 mg/kg/día de FA y 1.0 a 1.5 g de proteínas por kilo de peso, provenientes de la fórmula sin FA. Las calorías y otros nutrientes se ajustan según la edad y el peso trimestral del embarazo.
Es crucial medir el nivel de FA semanalmente, manteniéndolo por debajo de 4 mg/dL, y controlar la TIR plasmática mensualmente, asegurando que se mantenga sobre 0.8 mg/dL. Si los niveles de TIR son inferiores, se recomienda suplementar con 300-400 mg/día.
Fenilcetonuria - Video Explicativo
tags: #laboratorio #prinal #harina #pku #valores